GMP Anhang 1 – Auswahlkriterien für Reinraumschutzkleidung
Von Steve Marnach von DuPont.
Zum ersten Mal seit 2008 ist der Anhang 1 des EG-Leitfadens der Guten Herstellungspraxis (GMP) für sterile Medizinprodukte grundlegend überarbeitet worden. Der neue GMP-Anhang 1 wurde am 25. August 2022 veröffentlicht und ist mehr als ein einfaches Update. Der Leitfaden selbst wurde komplett überarbeitet und die Seitenanzahl von Anhang 1 nicht nur von 16 auf 58 Seiten erhöht, sondern auch der darin beschriebene Ansatz vollständig geändert. Daraus ergeben sich Konsequenzen sowohl für die Technologien als auch Verfahren der pharmazeutischen Herstellung.
Der folgende Auszug aus Abschnitt 2.2 auf Seite 4 beinhaltet eine Zusammenfassung des neuen Ansatzes:
„Prozesse, Ausrüstung, Einrichtungen und Fertigungsaktivitäten sollten in Übereinstimmung mit den QRM (Qualitätsrisikomanagement) -Grundsätzen verwaltet werden, die ein proaktives Mittel zur Identifizierung, wissenschaftlichen Bewertung und Kontrolle potentieller Qualitätsrisiken darstellen. (...) Die ausschließliche Überwachung oder Prüfung garantiert keine Sterilität.“
Es wird erwartet, dass alle Aktivitäten, die die pharmazeutische Herstellung betreffen, ganzheitlich durch die QRM-Grundsätze geregelt und in der Kontaminationskontrollstrategie (CCS) dokumentiert werden. Zudem wir gefordert, dass die Kontaminationskontrollstrategie (CCS) ein lebendes Dokument ist, basierend auf einem daten- und wissenschaftsbasierten Ansatz, der zur Kontrolle potentieller Qualitätsrisiken kontinuierlich aktualisiert und verbessert werden sollte. Der neue GMP-Anhang 1 fordert zu einem proaktiven Vorgehen auf. Das einfache Reagieren und Korrigieren von erkannter Kontamination wird nicht länger ausreichen. Es wird erwartet, dass Hersteller Ihre Prozesse und Verfahren vollständig verstehen, sodass sie potentielle Qualitätsrisiken identifizieren, alle technischen und verfahrensbezogenen Mittel, die zur Kontrolle dieser Risiken notwendig sind, umsetzen und kontinuierliche Verbesserungen anstreben können. Da Reinraumkleidungssysteme ein entscheidender Bestandteil der sterilen und aseptischen Herstellung sind, müssen sie gemäß den QRM-Grundsätzen gehandhabt werden.
QRM-Grundsätze für Reinraumkleidung
Das Qualitätsrisikomanagement (QRM) beginnt mit einer vollständigen datengestützten Analyse und Evaluierung aller Qualitätsrisiken in Verbindung mit Reinraummitarbeitern, die Reinraumkleidung tragen. Solch eine Bewertung erlaubt es dem Analysten, Zertifizierungs-, Qualifizierungs-, Validierungs- und Überwachungsverfahren als Qualitätssicherung zu entwerfen, die Teil einer ganzheitlichen Kontaminationskontrollstrategie (CCS) sind. Eine Risikobewertung ist erforderlich, um Kontaminationsrisiken bei Reinraummitarbeitern, die Reinraumkleidung tragen, zu verstehen. Reinraummitarbeiter sind die größte Kontaminationsquelle in Reinräumen und machen 75 % aller Kontaminationen aus.1,2 Diese Kontaminationen stammen sowohl von den Mitarbeitern selbst als auch ihrer Reinraumkleidung.
Die „menschliche“ Kontamination, verursacht durch Reinraummitarbeiter, entsteht sowohl durch ihre Biologie (ein Durchschnittsmensch verliert 40.000 Partikel pro Minute und 10 % davon transportieren Mikroorganismen) als auch ihr Verhalten. Während Letzteres durch die umsichtige Auswahl der Reinraummitarbeiter, Schulungen, langsame Bewegungen und korrekte Hygienemaßnahmen minimiert werden kann, bleibt es eine Tatsache (wie viele Studien zeigen), dass Reinraummitarbeiter Partikel abgeben. Die einzige Maßnahme zur Verhinderung von Reinraumkontamination durch Partikel von Reinraummitarbeitern ist das Tragen von Reinraumkleidung, die die einzige Barriere zwischen Reinraummitarbeiter und Produktionsumgebung darstellt. Der 2022 veröffentlichte GMP-Anhang 1 hebt diesen Umstand in Abschnitt 7.13 i hervor.
"Die Schutzkleidung sollte nahezu keine Fasern oder Partikel abgeben und vom Körper abgegebene Partikel zurückhalten."
Wie oben bereits erwähnt, stellt Reinraumkleidung als solche schon eine Kontaminationsquelle dar und muss als Risiko bewertet werden. So kann zum Beispiel das Material, aus dem die Bekleidung besteht (nicht gewebtes Material für Einwegbekleidung oder gewebtes Material für Mehrwegbekleidung), eine größere oder kleinere Anzahl an Partikeln abgeben, je nach Beschaffenheit der genutzten Fasern bzw. Fäden, der Abriebsbeständigkeit und der Konzeption. Auch die Verschlusstypen (Reißverschlüsse, Knöpfe, Gummizüge oder Nähgarn) können eine Kontaminationsquelle darstellen. Das Bekleidungsdesign kann somit ebenfalls eine Rolle spielen, was bedeutet, dass auch die Verschlusstypen bewertet werden müssen. Ein häufig vernachlässigtes Detail ist die Verpackung, in der die Reinraumkleidung geliefert wird, die eine zusätzliche Kontaminationsquelle sein kann (z. B. Papierbeutel versus Plastikbeutel).
Die wichtigsten Validierungsphasen
Ist die Risikobewertung einmal abgeschlossen, sollten die Risikoursachen so weit wie möglich mithilfe von technischen und organisatorischen Maßnahmen beseitigt bzw. ersetzt werden. Die Restrisiken sollten so gut wie möglich durch ein validiertes Reinraumkleidungssystem minimiert werden. Die EU-Grundsätze der Qualifizierung und Validierung (GMP-Anhang 1519) geben einen allgemeinen Rahmen vor, der zur Qualifizierung von Reinraumsystemen eingesetzt werden kann. Dieser Validierungsansatz besteht aus fünf Schritten: die Definition einer Benutzeranforderung (URS), einer Designqualifizierung (DQ), einer Installationsqualifizierung (IQ), einer Funktionsqualifizierung (OQ) und einer Leistungsqualifizierung (PQ). Obwohl die Designqualifizierung (DQ) und die Installationsqualifizierung (IQ) den größten Einfluss auf die erreichte Qualität haben, dürfen die anderen Schritte nicht vernachlässigt werden. Es ist wichtig, Schritt für Schritt vorzugehen.3
Benutzeranforderung (URS)
Obwohl sie formell nicht Teil des Validierungsprozesses sind, ist es wichtig, im Voraus die Anforderungen von Benutzer und Arbeitsumgebung an das Reinraumkleidungssystem zu definieren. Die Benutzeranforderung (URS) definiert die kritischen Anforderungen, anhand derer das Bekleidungssystem bewertet werden muss, damit es der Risikobewertung entspricht. Der geschulte Reinraummitarbeiter muss beispielsweise mindestens 3 Stunden in derselben Reinraumkleidung arbeiten können, ohne dabei eine Kontaminierung in inakzeptablem Umfang der Bekleidung und der aseptischen Arbeitsumgebung zu verursachen (gemäß aktuellem GMP (cGMP)). Das Verpackungssystem der Bekleidung muss für die Gegebenheiten des Reinraums und dessen Materialschleusensystem bzw. die manuelle Sprühdesinfektion geeignet sein. Manchmal braucht der Reinraummitarbeiter auch einen chemischen und biologischen Schutz vor Substanzen, mit denen im Reinraum gearbeitet wird.
Designqualifizierung (DQ)
Im Rahmen der Designqualifizierung (DQ) wird die Übereinstimmung des Reinraumkleidungssystems mit den Anforderungen des cGMP nachgewiesen und dokumentiert, um zu bestätigen, dass die ausgewählte Reinraumkleidung für den entsprechenden Verwendungszweck geeignet ist. Da der neue Anhang 1 einen daten- und wissenschaftsbasierten Ansatz voraussetzt, sollte die Designqualifizierung (DQ) Tests enthalten, die die entsprechende Verwendung und Leistung der Bekleidung simulieren. Wie durch ISO 11607-1 empfohlen, sollte die Designqualifizierung (DQ) in vier Schlüsselbereiche aufgeteilt werden: Materialqualifizierung, Leistungstests, Stabilitätstests und Eignungsprüfung. Für Mehrwegbekleidung muss der Prozess auf die Subunternehmer der Bekleidungshersteller, Lieferanten und Dienstleister ausgeweitet werden. PAVIČIĆ und WAGNER haben eine Liste der Eigenschaften erstellt, die bewertet werden müssen (Liste 1).3
Liste 1: Risiko- und wissenschaftsbasierte Validierung von Reinraumkleidung
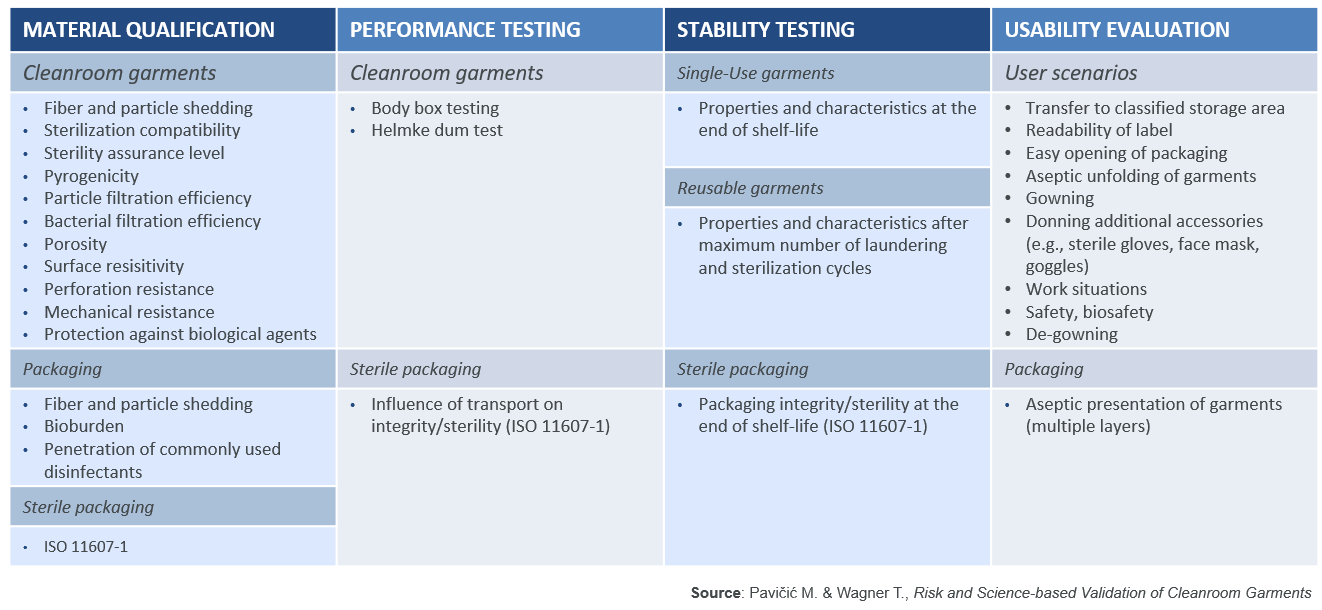
In diesem Artikel werden nur einige dieser Eigenschaften behandelt, um ihre wichtige Bedeutung zu zeigen, zusammen mit wissenschaftlichen Testmethoden, die genutzt werden, um die Leistung des Reinraumkleidungssystems zu bewerten.
Materialqualifizierung
Um zu prüfen, ob Kleidung wirklich steril ist, muss kontrolliert werden, ob der Hersteller einem validierten Sterilisationsprozess folgt, einen Sterilitätssicherheitsfaktor (SAL-Wert) von 10-6 (gemäß ANSI/AAMI/ISO 11137-1) garantiert und dies in einem Sterilitätszertifikat dokumentierten kann. Ein einfaches Bestrahlungszertifikat oder ein Dokument, das eine interne Dampfsterilisation bestätigt, ist nicht ausreichend.
Da Reinraumbekleidung eine Barriere für „menschliche“ Kontamination durch Reinraummitarbeiter darstellen muss, ist es wichtig, die Partikelfiltrationseffizienz der für diese Bekleidung genutzten Materialien zu bewerten (z. B. nicht gewebtes Material oder wiederverwendbares gewebtes Polyester). Die Partikelfiltrationseffizienz (PFE) gegenüber trockenen Partikel kann mit der Testmethode EN 143 (TSI 8130) bewertet werden, die die Filtrationseffizienz mithilfe von Salzpartikeln mit 0,3 µm Durchmesser misst. Die Bakterienfiltrationseffizienz (BFE) kann mit der Testmethode ASTM F2101 bewertet werden.
Leistungstest
Der Helmke-Drumtest (IEST-RP-C003.4) ist eine gute Möglichkeit, die Partikelemission von Reinraumkleidung zu bewerten, insbesondere für Bekleidung, die mehrere Male gewaschen wird. Unterdessen ist der Body-Box-Test (IEST-RP-CC003.4) die einzige Methode, um die Partikelemission bei realen Tragebedingungen durch den Reinraummitarbeiter zu bewerten. Dadurch können sowohl die Partikelemission der Bekleidung sowie deren Partikel- und Bakterienfiltrationseffizienz (PFE und BFE) unter Berücksichtigung der vom Reinraummitarbeiter abgegebenen Partikel beurteilt werden.
Stabilitätstest
Es ist wichtig, die Veränderung der Eigenschaften und Merkmale der Bekleidung zu prüfen, die durch Alter, Abnutzung und Anzahl der Reinigungs- und Sterilisationszyklen entsteht. Dazu müssen die Leistungsparameter, die oben aufgelistet sind, unter ungünstigsten Bedingungen validiert werden. Bei Einwegartikeln werden zum Beispiel Bekleidungsstücke aus verschiedenen Chargen und bei Ablauf der Haltbarkeit bewertet, während Mehrwegbekleidung nach 10, 20, 30, 40 und 50 Reinigungs- und Sterilisationszyklen bewertet wird, um ihre Haltbarkeit bestimmen zu können. Studien von ROMANO, LJUNGQVIST und REINMÜLLER haben belegt, dass wiederholtes Waschen die Leistung der Bekleidung verschlechtert.4,5

Eignungsprüfung
Es ist wichtig, Nutzungsszenarien durchzugehen und die Verpackung der Reinraumbekleidung zu bewerten, um zu gewährleisten, dass sie mit akzeptabler Restkontamination und akzeptablen Sicherheitsrisiken genutzt werden kann. Obwohl dies normalerweise durch die Endnutzer geschieht, können Lieferanten auch Prüfungen durchführen und den Nutzern Daten liefern.
Installationsqualifizierung (IQ)
Auch wenn die Installationsqualifizierung (IQ) der dokumentierte Nachweis ist, dass alle erforderlichen Elemente des Reinraumankleidesystems vorhanden sind, ist es wichtig, folgende Punkte zu prüfen, um unvorhergesehene Risiken auszuschließen:
- Sind Ankleide- und Entkleideeinrichtungen korrekt installiert?
- Hat der Lieferant die erforderlichen Konformitätszertifikate und/oder Analysen, Anweisungen usw. bereitgestellt?
- Wurden die Standardarbeitsanweisungen (SOPs) für Ankleide- und Entkleidesysteme erstellt oder angepasst?
- Wurden die logistischen Prozesse für Bekleidung und Zubehör validiert?
- Wurden Schulungs- und Qualifikationspläne für Reinraummitarbeiter erstellt?
Funktionsqualifizierung (OQ)
Die Funktionsqualifizierung (OQ) ist der dokumentierte Nachweis, dass das Ankleide- und Entkleidekonzept einschließlich Logistik und Materialschleusensystem sowie die aseptische Präsentation der Bekleidung (d. h. Faltung und Verpackung) funktionieren.
Leistungsqualifizierung (PQ)
Die Leistungsqualifizierung (PQ) wird normalerweise unter ungünstigsten Bedingungen abgeschlossen, die basierend auf einer Risikobewertung festgelegt werden. Ziel ist dabei die Leistungsvalidierung der Reinraumkleidungssysteme unter Realbedingungen. Die in der Benutzeranforderung (URS) aufgeführten Anforderungen müssen vollständig erfüllt sein. Sie schließen eine aseptische Ankleidequalifizierung und -validierung der mikrobiologischen Qualität des mit Reinraumkleidung und -zubehör eingekleideten Personals bei der tatsächlichen Arbeit mit ein. Natürlich endet der Prozess damit nicht: Die regelmäßige Revalidierung des Bekleidungssystems, konstante Überwachung und eine kritische Bewertung von Veränderungen in der Bekleidung oder dem Bekleidungssystem sind wichtig, um den Kontrollstatus zu belegen.
Zusammenfassung
Reinraumkleidungssysteme sind ein entscheidender Bestandteil der Kontaminationskontrollstrategie (CCS) und der Prozessvalidierung. Ein risiko- und wissenschaftsbasierter Quality-by-Design-Ansatz mit Verifizierung ist die richtige Strategie zur Kontrolle von menschbezogenen Kontaminationsrisiken und bietet integrierte Risikominderungen. Dieser Ansatz ist eine adäquate Antwort auf die neusten Vorgaben.
Quellenangaben
[1] Ramstorp M. (2000) Introduction to contamination control and cleanroom technology. Wiley VCH, Weinheim, Deutschland.
[2] Ramstorp M. (2019) Cleanroom garments from a quality risk management approach. Eur J Parenter Pharm 24(3):4–16.
[3] Pavičić M., Wagner T. (2019) Risk & science-based validation of cleanroom garments. IVT Network. https://www.ivtnetwork.com/article/risk-science-based-validation-cleanroom-garments
[4] Ljungqvist B., Reinmüller B. (2005) Aseptic production, gowning systems and airborne contaminants. Pharm Technol Suppl(2): https://cdn.sanity.io/files/0vv8moc6/pharmtech/75a6ab7e927edd600f5286a1eabb2b41171af658.pdf/article-160408.pdf
[5] Romano F., Ljungqvist B., Reinmuller B. et al. (2016) Performance test of technical cleanroom clothing systems. Proceedings of Indoor Air 2016, 14th International Conference on Indoor Air Quality and Climate, Ghent, Belgien.

Inhalte bereitgestellt durch:




